Blog on CRISPR technology
What you need to know about the CRISPR technology as a potential therapy for DMD
- CRISPR technology (or genome editing) is a technique that can change DNA at specific locations
- For Duchenne, genome editing can in theory bring about permanent exon skipping; so far this has been shown possible in cell models and a subset of muscle fibers of mdx mice
- Genome editing will only have a therapeutic effect if a large number of muscle cell nuclei are corrected in patients – at the moment this cannot be achieved in humans
- The tools for genome editing need to be delivered to the body with viral vectors (similar to gene therapy); it is not yet possible to treat all the muscles in the human body with viral vectors
- Genome editing is a new technique and more optimization is needed. The current tools are not 100% specific, thus risking the introduction of mutations in other genes
- Genome editing is not a cure for Duchenne: muscle tissue and function that is lost will not return
- Genome editing will most likely not be in clinical trials for DMD in the near future!
- Delivery of genome editing tools (gene therapy) needs to be optimized (something the gene therapy has worked on for decades, so far with only limited success)
- The safety aspects of genome editing need to be charted
If you want to read the complete explanation of how CRISPR works and what the challenges are exactly, please continue reading.
Why you should not go along with the CRISPR cure hype
You’ve heard about CRISPR and genome editing and it is amazing potential and how it may be a cure for Duchenne in as little as 3 year. While I would love for this to happen, I do not think this is a realistic picture. And while I do not want to dash your hopes, I do believe there are some things you need to be aware off so you can have more realistic expectations.
How does genome editing CRISPR work and why is it so hyped?
Let’s start out with some background on how genome editing works and how CRISPR comes into play in this. Our genes consist of DNA and contain the genetic code for proteins. When there are mistakes (mutations) in genes, this will often result in the absence of functional proteins or the production of toxic proteins and lead to disease (Figure 1). Genome editing aims to modify the DNA, to correct the mistake and allow the production of a missing protein or a reduction of toxic protein production.
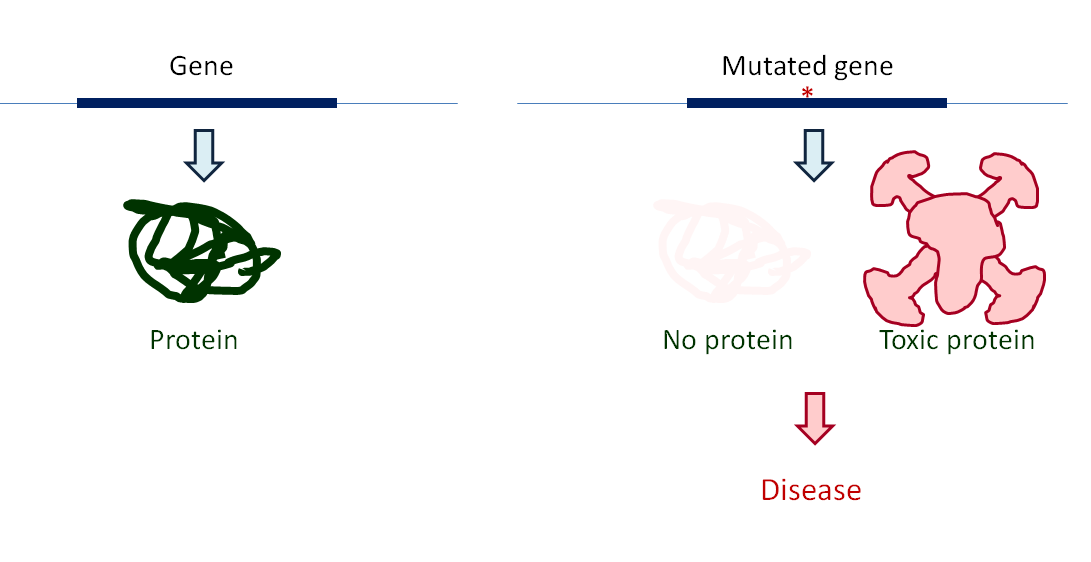
You need tools to modify DNA. Genome editing makes use of the systems in the cell that repair DNA when it is damaged. These systems are activated when the DNA is damaged (e.g. UV light can generate breaks in the DNA of your skin cells). When breaks occur in the DNA, the DNA repair systems are immediately activated, because DNA will quickly ‘unravel’ when it is broken and the cell does not want to lose genetic information.
In order to utilize the repair system for genome editing, one needs to activate the repair system by generating a break in the DNA at the location of the mutation. This is where CRISPR comes in – CRISPR is a tool that recognizes specific locations in the DNA and guides an enzyme that can cut the DNA (Cas9) to this location. The beauty of the CRISPR is that one can design the DNA recognizing component (“guide”) to recognize specific parts of the DNA. While there are some rules that have to be adhered to, it is possible to design these components to specifically target most regions in the genome. Thus it now becomes possible to cut at or close to mutations.
There are two repair pathways that can be activated, called non-homologous end-joining (NHEJ) and homologous recombination (HR). NHEJ glues the two ends together – however, because DNA often unravels a little bit before the break is repaired, often some of the DNA is lost in this process. HR uses a template and exchanges this for the part that is broken (Figure 2). So here there is no loss of DNA. With this system, you can correct mutations: when you introduce a break close to the mutation and provide a template with the correct DNA this template will be used to repair the break and the mutation is corrected. Unfortunately, the HR system is very inefficient and does not occur at all in non-dividing cells like muscle. So for muscle diseases only the NHEJ system can be used for now.

How does CRISPR work for Duchenne?
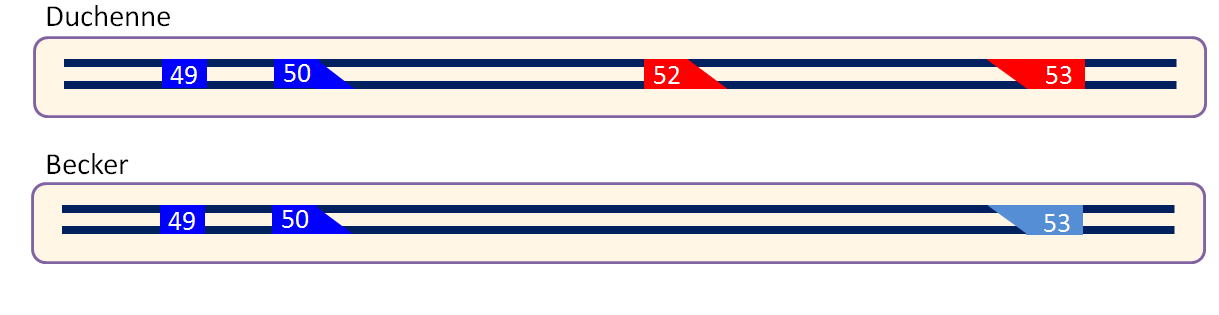
As explained, in muscle tissue only the NHEJ process is active. The NHEJ process results in minor mutations in the repaired DNA. This is generally not something that is desired. However, for DMD this loss of DNA can be exploited to restore the genetic code of the dystrophin gene. As you are probably aware, DMD is caused by mutations in the dystrophin gene that disrupt the genetic code. For most genes, including dystrophin, the genetic code lies dispersed over the gene in so called exons. Most DMD patients miss one or more exons, causing the genetic code to be disrupted (Figure 3). When patients miss exons without disrupting the genetic code, a partially functional dystrophin can be produced. These patients have a less severe disease called Becker muscular dystrophy.
The exon skipping approach aims to restore the genetic code by deleting an extra exon on transcript level. The disadvantage of this system is that transcripts are temporary gene copies with a high turnover, and therefore patients need to be treated with the exon skipping compounds regularly (weekly in current clinical trials). If it would be possible to delete an extra exon on DNA level, this would result in a permanent effect: all transcripts produced from the DNA would have a readable genetic code.
The CRISPR technology has been exploited in several ways to delete an exon – the two most commonly used are explained in Figure 4. The first system uses two guides to target a break at each side of the exon. When the repair system repairs this break it will sometimes join the two outer ends, so the whole exon is deleted. This restores the genetic code on DNA level.
Another option is to use only one guide and generate a single break close to the border of the exon. This is where exon recognition signals are located. When the break is repaired, some DNA subunits will be lost in the process, and consequently the exon recognition signals will be lost. So while the exon is still present in the DNA, it is no longer recognized by the cellular machinery and will not be included on transcript level. Now the genetic code is restored on transcript level.

Permanent exon skipping! That sounds amazing. So what is the catch?
The catch is that you need tools to introduce the breaks: the Cas9 enzyme that generates the DNA breaks and the CRISPR guide(s) to lead the Cas9 enzyme to the proper location in the DNA. It is not enough to have these tools in one nucleus of one muscle fiber – the tools need to be delivered to a significant number of nuclei in muscle fibers of each of the more than 750 different muscles of the human body – because almost all of these are affected by Duchenne. How can we do this? Antisense oligonucleotides (AONs) that are used for exon skipping are small and will be distributed throughout the body and take up by muscle fibers to some extent. However, when you inject the Cas9 enzyme, it will not be taken up by muscle fibers. The trick that is currently used is to generate a viral vector containing the genetic information for the Cas9 enzyme and another one containing the genetic code for the guides. The adeno-associated virus (AAV) can used to deliver these genes to muscle (AAV can infect muscle with reasonable efficiency). AAV vectors have been used to deliver Cas9 and guides to muscle of Duchenne mouse models.
Indeed! There are three papers in Science showing that CRISPR works in DMD mouse models. So the proof has been provided and we should start planning a clinical trial!
Unfortunately, things are not that simple. Gene therapy been shown to work in DMD mouse models for decades (viral vectors delivering a custom made gene encoding a mini-version of dystrophin). However, it has not yet been possible to treat all the muscles of a human with viral vectors. Gene therapy for muscle diseases faces multiple challenges and most of these challenges are also faced by the CRISPR system:
- We have a lot of muscle (~30-40% of our body is muscle), this means that a lot of viral vector particles are needed to treat a good amount of muscle fibers in each of the 750 muscles
- Muscle tissues does not take up viruses that well. It is possible to increase the uptake of viruses for an arm or a leg using high pressure (hydrodynamic limb perfusion) – if humans respond the same way as dogs, this is anticipated to lead to good uptake of the viruses in one arm or leg and some uptake in the rest of the body
- Treatment with viral vectors will illicit an immune response (the immune system does not know that these virus has good intentions)
- The AAV virus does not integrate into the DNA. In healthy muscle it will be stable for ~10 years. However, in dystrophic muscle the turnover is higher. Work in a Duchenne dog model has shown that the virus and the transgene are lost after ~5 years. For the CRISPR system this is less of an issue than when AAV is used to deliver a mini-dystrophin gene, because once the mutation has been corrected, the CRISPR system is no longer needed. However, on the other hand, the CRISPR system is a two-tiered system: the Cas9 tools AND the guides need to be delivered to the same nucleus in order to work. Then they also have to do their job (the cutting) and the cell will have to do its job (the repairing). These events will not happen in all the targeted cells but only for a subset. So the CRISPR system is expected to be less effective than gene addition therapy where all nuclei that were targeted can produce the mini-dystrophin protein
- Like all therapeutic approaches, gene therapy relies on muscle quality. Dystrophin prevents muscle damage during exercise. Once muscle has been lost, it will not return. Therapeutic approaches currently in development for Duchenne aim to slow down disease progression, they will not bring back muscle that is lost – they are not cures. Genome edition is no exception to this. When disease progresses in Duchenne patients, muscle tissue is replaced by fat and adipose tissue. Fibrotic and adipose tissues do not produce dystrophin, so repairing the dystrophin gene in these tissues will not have an impact – only repairing the dystrophin gene in muscle tissue will.
Are there risks involved?
As with most therapies, there are risks and potential side effects. Genome editing however has an added risk in that it targets DNA, so the effects are permanent and will not go away when treatment is stopped. The genome editing system is not fool-proof, CRISPR has been known to direct Cas9 also to other locations than the target, leading to cuts in the DNA at the wrong location. There are multiple ways in which this can leads to problems, for example this can lead to cells being unable to produce proteins that prevent tumor formation. As said, the effects in the DNA are permanent and cannot be undone. There is no undo button with genome editing.
So what needs to be done?
I am not saying this is the end of all hope for genome editing for Duchenne. I am pointing out that we need to be cautious. We should not expect too much from this approach (it is not a cure, it slows down disease progression) and we should not expect things too soon. Before clinical trials can be started, the delivery of the tools needs to be optimized. This should not be underestimated. The step from a 25 gram mouse with 16 gram muscle to a 30 kg Duchenne boy with 20 kg muscle is huge. Furthermore, more information needs to be obtained on the risks of this technology – not just for Duchenne, but in general: How serious are the risks? Can they be mitigated? Do they outweigh the risk of having Duchenne? This will take time – something I know Duchenne patients do not have. However, I know from personal experience that rushing into clinical trials seldom turns out well and leads to further delays.